Lig3DLens: An open source ligand-based 3D virtual screening toolbox
Dall-e prompt: Navigating the chemical space
Why?
A common computational task we perform in drug design involves exploring the chemical space around an ‘initial’ hit molecule. Several commercial packages, like OpenEye, Cresset, and Schrödinger, are available for this purpose. Recently, an open-source alternative called VSFlow [1] has been introduced, which facilitates the preparation of a commercial compound database and provides three different ligand-based virtual screening modes.
Here at Healx, we have developed an end-to-end computational toolbox for 3D virtual screening based on the shape and electrostatics similarity to a reference (hit) compound. A drug designer can use a reference molecule and a (commercial) compound library of interest as inputs. The final output is a set of chemically diverse compounds with high 3D similarity to the reference compound.
What?
In Lig3DLens (L3DL) we are employing RDKit [2] to generate the 3D conformers of the library compounds and align them to the reference compound. We use the ESPSim package [3] to include an electrostatics term in the 3D scoring function. Additionally, we provide a tool for clustering the top-ranking hits obtained from 3D virtual screening (VS) and selecting a final set of compounds to purchase and test.
L3DL consists of three modules: i) preparing a commercial library that will be used as the chemical atlas, ii) generating the 3D conformers and overlaying them to the reference molecule using one of RDKit’s shape alignment method (rdMolAlign) [4], iii) clustering the top ranking compounds and selecting a predefined number of compounds that can be ordered and tested in an assay relevant to the project.
Please note that this technical walk-through focuses on the code’s functionality, and we do not delve into theoretical insights behind the methods employed in this computational toolbox.
Lig3DLens’ open source code can be found here.
How?
1. Compound library preparation
At this step, two main tasks are performed: i) Standardising the chemical structures in the compound library and ii) filtering compounds based on predefined ranges in the physicochemical property space. For the pre-processing of molecular structures, we employ the datamol [5] package. The user can provide input files in sd, csv, or ascii formats, which should contain the SMILES and IDs of the compounds. Additionally, the code allows to filter out compounds with undesired physicochemical properties. The ranges for these properties can be defined in the input yaml file. The resulting output is a sd file containing the 2D structures and any ID columns found in the input file.
lig3dlens-prepare --in input_SD_file \
--filter physchem_yaml_file \
--out output_SD_file
For this test VS run we used the Enamine hit locator library [6] and a Wee-1 inhibitor (ZN-c3)[7].
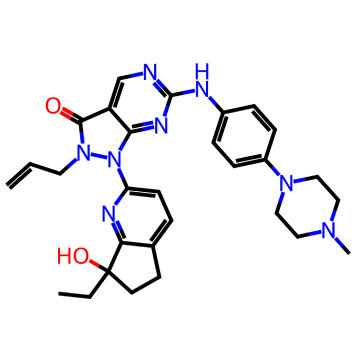
Figure 1. Azenosertic (ZN-c3) from Zendalis Pharmaceuticals. 2D and 3D representations of ZN-c3.
lig3dlens-prepare --in Enamine_Hit_Locator_Library.sdf \
--filter physchem_properties.yaml \
--out Enamine_Hit_Locator_Library-filtered.sdf
The script takes approximately 30 mins to standardise the chemical representations of the compounds in the commercial library, calculate the physicochemical properties for all the compounds in the dataset, filter based on the criteria defined in the .yaml file and store the final set of compounds to a SD file. The Enamine hit locator library contains ~ 460,000 molecules and approximately 22,000 compounds are removed from the initial set after applying the physicochemical properties filters. The physicochemical properties space of the filtered set (~ 438,000 compounds) is represented in Figure 2.
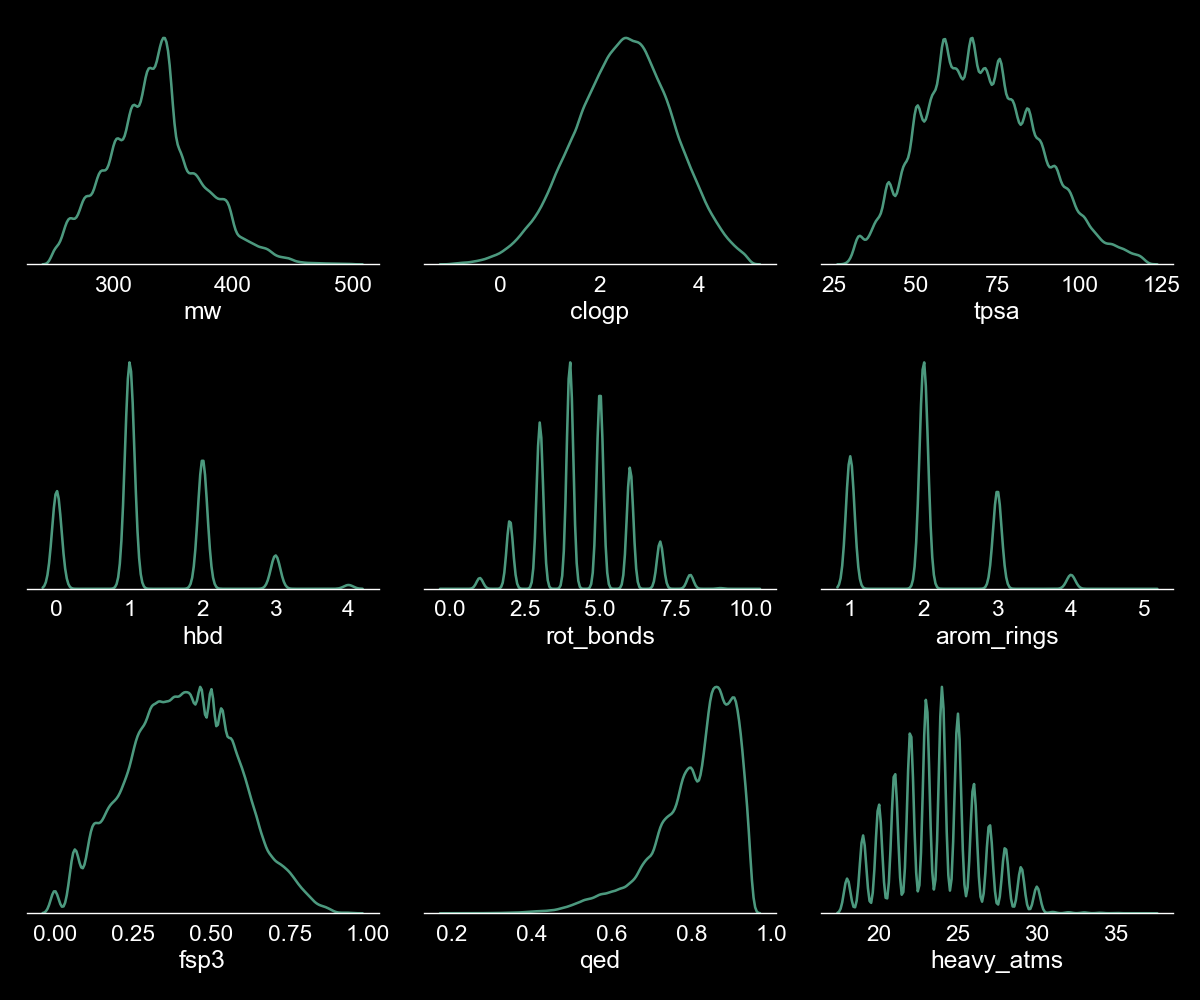
Figure 2. Physicochemical space of the filtered Enamine library. We used the kdeplot function from the seaborn package to plot the above figure. The y-axis represents the density of the depicted physicochemical properties and has been omitted from these subplots.
2. 3D shape and electrostatics similarity
In this step we score a set of compounds based on their 3D similarity to a reference compound (initial hit molecule/starting point). In order to do that we generate the 3D conformers for both the library compounds and the reference molecule using RDKit. The user can define the number of conformers for the library compounds, whereas for the reference compound we generate only one conformer.
For the conformer generation, we are using RDKit (AllChem.EmbedMultipleConfs) and it is performed in parallel using almost all of the available cores. The parallelisation of this step has been inspired by Pavel Polishchuk’s codebase. For the 3D shape alignment of the library compounds to the reference molecule we use rdMolAlign function. Also, we calculate the 3D shape and electrostatics similarity of a library compound to a reference molecule using the
ESPSim package, by default we use the Gasteiger charges for the electrostatic similarity calculation.
The final output file includes the best 3D overlay of the library compound with its ID columns, the shape, electrostatic and feature map scores. This step of the workflow is the most demanding in computing resources and it takes approximately 11 hours to complete in a m5.2xlarge AWS instance (8 vCPUs, 32 GB RAM).
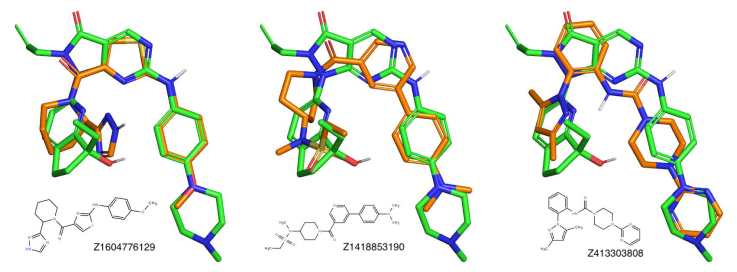
Figure 3. 3D overlays of three representative compounds to the reference molecule (ZN-c3). We used the open source versions of Pymol and Ketcher for the 3D and 2D molecular representations.
lig3dlens-align --ref input_reference_molecule_file \
--lib input_library_file_name \
--conf num_conformers \
--out output_SD_file
lig3dlens-align --ref reference_mol.smi \
--lib Enamine_Hit_Locator_Library-filterd.sdf \
--conf 10 \
--out Enamine_Hit_Locator_Library-VS_results.sdf
3. Compound selection (clustering)
For this case study, we've extracted the top 10,000 ranked compounds from the preceding step in our workflow, which involved 3D virtual screening. Our objective is to identify 500 compounds that are readily available from commercial sources for subsequent testing in our assays. The exact number of compounds to be tested sometimes can be determined by our budget constraints and/or the throughput of our assays.
To achieve the selection of these 500 compounds, we employ the k-means clustering algorithm, drawing on elements from Pat Walters' codebase. This step involves clustering the highest-scoring compounds into a user-defined number of groups, followed by the identification of representative members within these clusters.
The code accepts as an input a SD file containing the highest scoring compounds, along with parameters like the desired number of clusters, the preferred type of fingerprints, and their designated length. The code’s main output is a CSV file containing the SMILES representations of compounds and the corresponding IDs for the cluster centres.
We employ the molfeat package to streamline the fingerprint generation process.
lig3dlens-cluster –-in input_SD_file \
–-clusters num_clusters \
–-out output_file \
–-fp_type fingerprint_type \
–-dim fingerprint_dimension
lig3dlens-cluster --in top10K_VS_results.sdf \
–-clusters 500 \
–-out top10K_VS_results-clusters.sdf \
–-fp_type ecfp \
--dim 1024
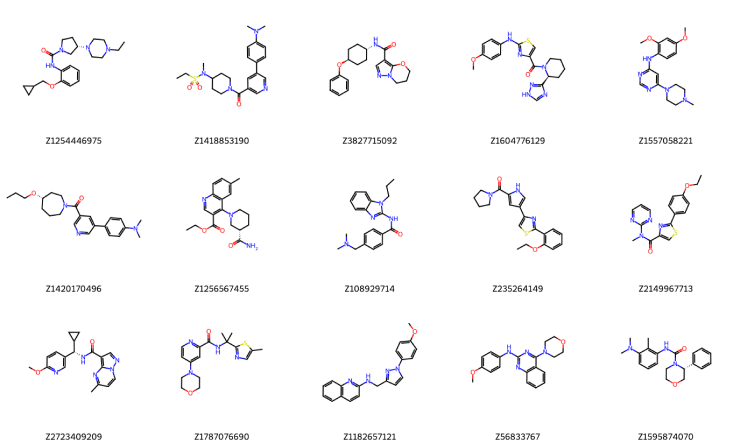
Figure 4. A few representative examples out of the final selection set (500 compounds). We use the Enamine ID as the compound's legend.
References
1. Jung S.; Vatheuer H.; Czodrowski P. VSFlow: an open-source ligand-based virtual screening tool. Journal of Cheminformatics, 2023, 15:40.
3. Bolcato G.; Heid E.; Böstrom J. On the value of using 3D shape and electrostatics in deep generative models. Journal of Chemical Information and Theory, 2022, 62, 1388-1398
4. http://www.rdkit.org/new_docs/source/rdkit.Chem.rdMolAlign.html
6. https://enamine.net/compound-libraries/diversity-libraries/hit-locator-library-460
7. Huang P.Q.; Boren B.C.; Hedge S.G.; Liu H. Unni A. K.; Abraham S.; Hopking C.D.; Paliwal S.; Samatar A.A.; Li J. Bunker K.D. Discovery of ZN-c3, a Highly Potent and Selective Wee1 Inhibitor Undergoing Evaluation in Clinical Trials for the Treatment of Cancer, Journal of Medicinal Chemistry, 2021, 64 (17), 13004-13024
Acknowledgements
We would like to thank the open source community for providing some of the tools that helped us compile this 3D virtual screening toolbox. More specifically, we would like to thank the RDKit community, Esther Heid (ESPSim), Pat Walters (Relay Tx), datamol/molfeat team from Valence labs, and Pavel Polishchuk.
